 |
|
 |
|
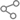
2月28日是国际罕见病日。目前,我国的罕见病患者超2000万人。罕见病80%由基因缺陷导致,50%于儿童期发病,5岁以内死亡率高达30%。上海市罕见病诊治中心、上海市儿童罕见病诊治中心挂牌于上海交通大学医学院附属新华医院,多学科专家组建成为一支专业且强大的罕见病诊治团队,为患者撑起了一片天。

图说:新华医院儿内分泌遗传科主任医师邱文娟 新民晚报记者 徐程 摄(下同)
诊疗方式改变,多学科专家围着患者转
1岁半的美美活泼可爱,但一年多来,她的爸爸妈妈很少体会到为人父母的喜悦。因为,他们有个不能说的“秘密”。出生时,美美的生殖器发育异常,但外阴像女性,父母一直把她当女孩抚养。随着年龄增长,美美腹股沟处发现了两个包块,当地医生觉得像男孩子的睾丸。不知美美是男是女,眼看还有一年就要进幼儿园了,爸妈很担心,于是带着孩子来到新华医院就诊。经B超检查发现,美美的双侧腹股沟的确存在睾丸,染色体分析显示她携带男性Y染色体。
新华医院儿内分泌遗传代谢科主任邱文娟给美美做了外显子测序,根据检测结果她告诉美美的父母,孩子得的是一种常染色体隐性遗传病——5α还原酶缺乏症,这是一种雄激素合成异常疾病,虽然现在外生殖器看起来像女孩,但青春期后这样的孩子可出现明显的男性青春发育和男性第二性征,到时候仍将面临性别选择的问题。
到底是做男孩还是女孩?最好早点决定下来。为此,邱文娟为家长预约了多学科门诊,邀请了儿童泌尿外科、遗传中心、伦理委员会等相关领域专家一起坐下来评估讨论。
如果选择做女性,孩子无需改换性别,但将来缺少子宫、卵巢等女性生殖器官将无法生育;如果选择做男性,接下来的治疗中,将接受双氢睾酮治疗和生殖器矫形手术,并需要将抚养性别和社会性别改为男性。“性别选择越早越好,这对孩子的性别意识养成和社会适应都有好处。5α还原酶缺乏症国际上还是建议当男性抚养,最好早点把孩子的社会性别改过来。”专家说。最终,家长听从了建议。
在邱文娟和罕见病患儿打交道的这么多年里,她见证了无数家庭与疾病斗争的痛苦。“最痛心的,是罕见病患儿错过最佳治疗时期。最欣慰的,是他们愿意跟随我们一步步治疗,最终控制住了疾病。”邱文娟说,过去传统的模式是一个患者挂号看不同的学科,辗转多年也不一定能得到诊断;现在多学科医生和专家围绕疾病、围绕患者,诊疗水平也大幅提升。

图说:邱文娟正在病房查房
无法治愈,并不等于无法干预
“罕见病多为遗传病,由于基因缺陷,确实无法实现治愈,但不代表无法治疗和干预。有很多罕见病通过积极治疗,患者可以回归社会,过上正常的生活。”邱文娟说。
一对姐弟都患罕见的代谢性疾病——多巴反应性肌张力不全。3年前,孩子父母带着儿子找到邱文娟就诊。他7岁了,走路没有力气,更别提跑跳了。家里人一度怀疑孩子也会像12岁的姐姐一样逐渐变得虚弱、瘫痪在床。幸好,新华医院让男孩获得明确诊断。经药物治疗,弟弟恢复很好。治疗弟弟的同时,邱主任想起了他的姐姐,她脊柱严重侧弯,行动无法自理,甚至连上厕所、穿衣这样的简单动作都需要妈妈帮忙。姐姐其实也是同样的病,只是她耽误了太久。
“这个家庭生育了一个有病的孩子后,就想再生一个。夫妻俩从未考虑过这可能是遗传的罕见病,没有做基因检测,谁知道两个小孩都是这样。”邱文娟说,后来医院为姐姐联系了小儿骨科的杨军林主任,一次脊柱手术后,这个脊柱侧弯超过40度、终日卧床的孩子坐起来了;一年后,杨旋主任给她做了马蹄内翻足的手术,她可以撑起支具独自挪出了家门;6个月后又做了髋关节的手术……看着女孩愈来愈自信的笑容,医生们都很高兴,“我们希望她的人生轨迹就此改变!”
在邱文娟看来,诊治罕见病的医生,不仅考验抽丝剥茧的审慎,更有不畏疑难的执着,以及陪伴患者一起走下去的信念。一句“医生会陪着你”,有时比药更让患者坚定。
一位经历颇为坎坷的成年女性尿素循环障碍患者,平日是要限制蛋白质摄入的。她25岁怀孕时发病进了新华医院重症监护室才获得了诊断和治疗。孕期,她为了补营养,激发了高氨血症发作,虽然命救回来了,孩子生出来也遗传了妈妈的基因,不久便夭折了。第二次怀孕时,她在新华医院做羊水穿刺,发现腹中胎儿还是该突变基因的男性半合子。崩溃,大哭,她放弃了这个孩子。第三次怀孕,她选择了第三代试管婴儿,也就是在胚胎植入前就进行遗传学诊断筛查,选出不带致病基因的健康胚胎植入。孕期虽然一路波折,但有了新华医院的危重孕产妇抢救中心保驾护航,她最终诞下一名健康的孩子。
管好一个病人,也管好一个家族
多年经验的积累,让新华医院逐渐探索出一个覆盖孕前至成人期的罕见病全生命周期诊疗体系,并成为全国第一批罕见病诊疗协作网中上海市的省级牵头医院。

图说:肾脏风湿免疫科副主任医师林芙君
“我们有罕见病初筛门诊、各学科的罕见病专病门诊,如遇疑似疑难罕见病或诊断不明疑似罕见病,医院还会启动罕见疑难病多学科(MDT)会诊,与患者共同寻找治疗的方向和方法。医院打造了一个可以诊断罕见病的院内专家库,只要有需求,都可以预约专家库的医生来会诊,他们具有丰富的罕见病诊断经验。”新华医院上海市罕见病诊治中心秘书、肾脏科副主任医师林芙君说。
不过,随着医疗技术的不断进步,一些罕见病患儿经过治疗,已长大成人,这些患者迫切希望能够无缝转诊至成人相关科室继续接受治疗。如何打通这一治疗断层?2021年,新华医院上海市罕见病诊治中心与复旦大学附属儿科医院徐虹教授团队共同启动了遗传及罕见病家庭防控多学科诊疗门诊,让患儿家庭中的患病亲属能在同一诊疗单元实现共同就诊。
林芙君的病人中,有不少是一大家子五六个病人一起来看病。30多岁的女性患者小王因为出现了血尿到肾脏科就诊,经仔细询问,林芙君发现,小王的父亲、大伯、大伯的儿子都有肾脏疾病,其中大伯已诊断尿毒症,做血透十多年。
这个家族患有典型的遗传疾病——Alport综合征,由于基因突变导致,临床表现以血尿、蛋白尿及进行性肾功能减退为特点。Alport综合征目前还没有根治办法,现阶段采取的是对症治疗。
经过基因检测发现,小王的女儿也未能幸免,只不过症状较轻。后来,一家子便选择家庭防控多学科诊疗门诊,一起随访。“家庭一起来,而且一家人的生活习惯和环境相似,更便于自我管理。”林芙君说。
期待更多新技术新药物出现
当然,林芙君也偶尔会感受到矛盾。她曾遇到一个18岁男孩,同样被诊断为Alport综合征。男孩在网上搜索发现这个疾病的最终归宿是尿毒症后,情绪非常低落,求学升造计划也取消了。其母亲经基因检测同样也是该病患者,陷入了深深自责。母亲对医生产生了抵触情绪:“你为什么要把我们诊断出来?我们一家子都被你毁了。”
“人们往往认为所有遗传性肾病都会以尿毒症为结局,但这不是绝对的,甚至同一个家系携带同一个位点的患者临床结局也可能天差地别。很多人不愿意接受患病的事实,他们内心恐惧,有时需要在门诊花一个小时甚至更多时间,来讲清楚疾病。”光是Alport综合征的来龙去脉,她就详细解释过上百次。
这几年,国内的罕见病诊疗水平、药物准入、医疗保障等都有了巨大的改观,但是,目前对于大部分罕见病我国还没有非常详实的流行病学数据。“中国有哪些罕见病病种,有多少人,分布在哪里?各个疾病的表型谱和基因突变谱的特点是什么?有效治疗方法是什么?标准诊断流程该是什么?这些都是亟待回答的。”林芙君告诉记者,几年前北京牵头建立国家罕见病注册登记平台,就是想解决这些问题,形成一批高质量的罕见病队列。
2019年,国家卫健委在国内组建罕见病诊疗协作网,推动国内罕见病诊疗水平的发展。社会各界对于罕见病的关注已越来越多,尽管诊断难、药物少的境况仍不易改,但在开放的交流下,新政策、新药物、新技术一定会不断突破现状,最终都指向患者对治疗方案的可及。
新民晚报记者 左妍
新民报系成员|客户端|官方微博|微信矩阵|新民网|广告刊例|战略合作伙伴
北大方正|上海音乐厅|中卫普信|今日头条|钱报网|中国网信网|中国禁毒网|人民日报中央厨房
增值电信业务经营许可证(ICP):沪B2-20110022号|互联网新闻信息服务许可证:31120170003|信息网络传播视听节目许可证:0909381
广电节目制作经营许可证:(沪)字第536号|违法与不良信息举报电话15900430043|跟帖评论自律管理承诺书
 |沪公网安备 31010602000044号|沪公网安备 31010602000590号|沪公网安备 31010602000579号
|沪公网安备 31010602000044号|沪公网安备 31010602000590号|沪公网安备 31010602000579号
新民晚报官方网站 xinmin.cn ©2023 All rights reserved



